









")


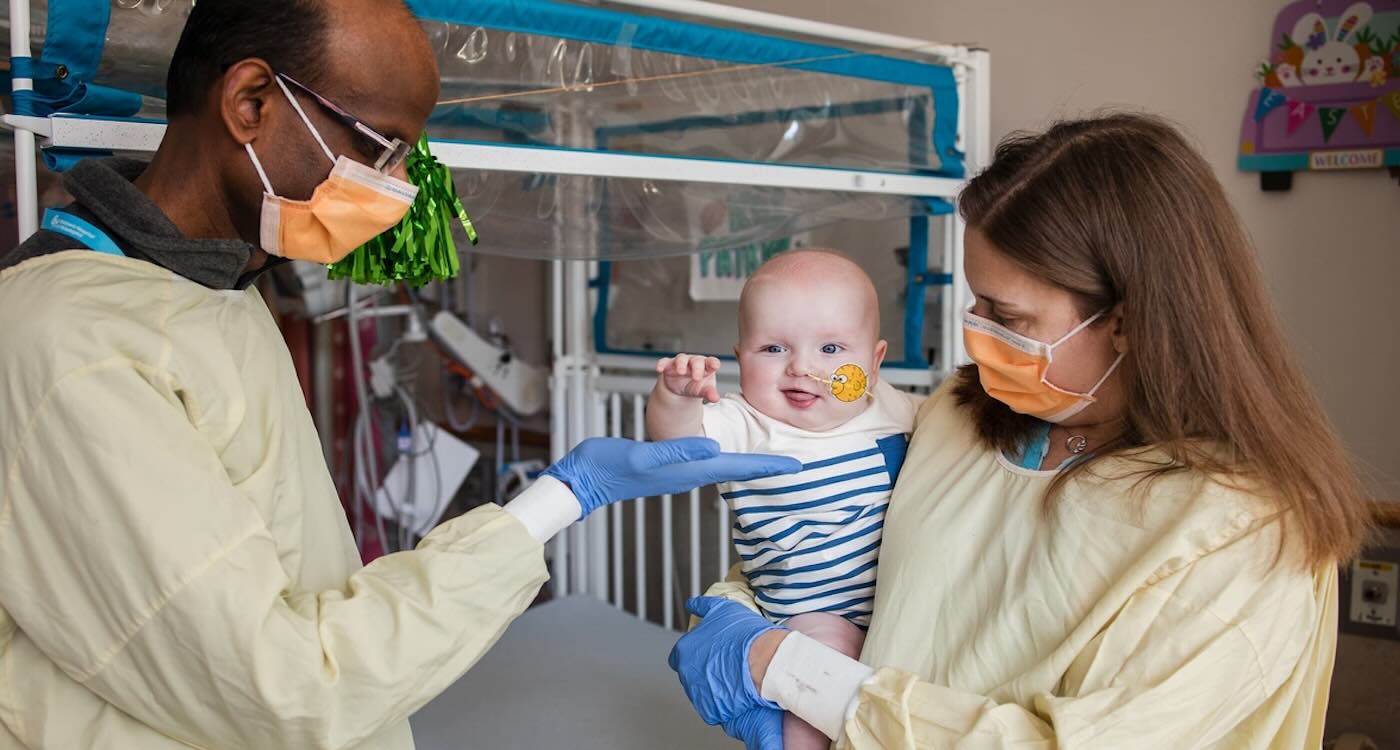

CRISPR has been used to create a genetic therapy option for a child born in Pennsylvania with a rare metabolic disorder.
Unable to convert ammonia to urea, newborn KJ was in serious risk of brain or liver damage, and had to be kept on medications and an extremely restrictive diet to avoid protein metabolism.
Children’s Hospital of Philadelphia (CHOP) doctors believed they could use CRISPR to develop a treatment to correct a faulty gene in KJ’s genome that would essentially cure him.
KJ’s parents, Nicole and Kyle Muldoon, decided to place their son’s wellbeing in the hands of two pioneering genetic therapists, Dr. Rebecca Ahrens-Nicklas and Dr. Kiran Musunru, who crafted a bespoke treatment that has successfully corrected the genetic defect.
“Years and years of progress in gene editing and collaboration between researchers and clinicians made this moment possible, and while KJ is just one patient, we hope he is the first of many to benefit from a methodology that can be scaled to fit an individual patient’s needs,” said Rebecca Ahrens-Nicklas, MD, PhD, director of the Gene Therapy for Inherited Metabolic Disorders Frontier Program (GTIMD) at Children’s Hospital of Philadelphia.
She, along with Dr. Musunru, are members of the NIH-funded Somatic Cell Genome Editing Consortium, and have spent years developing the science of using CRISPR to create individual treatment doses for the rarest of diseases.
So far, the only FDA-approved and standardized CRISPR therapies target two diseases found in tens of thousands of patients. CRISPR is an incredibly complex tool and expensive to wield, leaving its magic beyond the reach of millions of children and adults worldwide who collectively suffer from extremely rare genetic disorders.
One such disorder is called severe carbamoyl phosphate synthetase 1 (CPS1) deficiency, and it creates the inability to properly convert ammonia into urea to be excreted through urine. Ammonia is created in the body through protein metabolism. CPS1 is created in the liver to turn it into urea so as to avoid the toxic effects of the former.
KJ’s body cannot, so excess protein metabolism creates a buildup of ammonia in his liver that could be fatal. Nitrogen scavenging medication and a protein-deficient diet can keep a patient going until a liver transplant can be found, but at just months old, KJ’s body isn’t capable of enduring the procedure.
MORE RARE DISEASE RECOVERIES: UPDATE Long-Term Follow-up in Babies Born with ‘Bubble Boy Disease’ Still Seem Cured
A news release from CHOP reports that Ahrens-Nicklas and Musunuru targeted KJ’s specific variant of CPS1 after years of work with similar disease-causing variants. Within 6 months, their team designed and manufactured a base editing therapy delivered via lipid nanoparticles to the liver in order to correct KJ’s faulty enzyme.
In late February, 2025, KJ received his first infusion of this experimental therapy, and since then has received follow-up doses in March and April 2025, the release details. In the newly published New England Journal of Medicine paper, the researchers, along with their academic and industry collaborators, describe the customized CRISPR gene editing therapy that was rigorously yet speedily developed for administration to KJ.
WHISPER ABOUT CRISPR: Always Fatal Huntington’s Disease is Successfully Treated for First Time With Gene Therapy
KJ has received 3 doses, and suffered no side effects. He’s been able to halt medication and work some protein back into his diet, though he will need careful monitoring the rest of his life.
“We thought it was our responsibility to help our child, so when the doctors came to us with their idea, we put our trust in them in the hopes that it could help not just KJ but other families in our position,” his mother, Nicole, told CHOP.
SHARE This Incredible Recovery Story For A Baby In Serious Danger…
26 years ago, we struggled to map the human genome. Now, we are using our genomic knowledge to cure previously uncurable diseases.